Non-targeted analysis using LC-MS/MS DDA acquisition¶
This tutorial walks you through the workflow for analyzing non-targeted liquid chromatography high resolution mass spectrometry (LC-MS/MS) data-dependent acquisition (DDA) data starting from input file generation, to processing the data in SmartPeak, to reviewing the data in SmartPeak, to reporting the results.
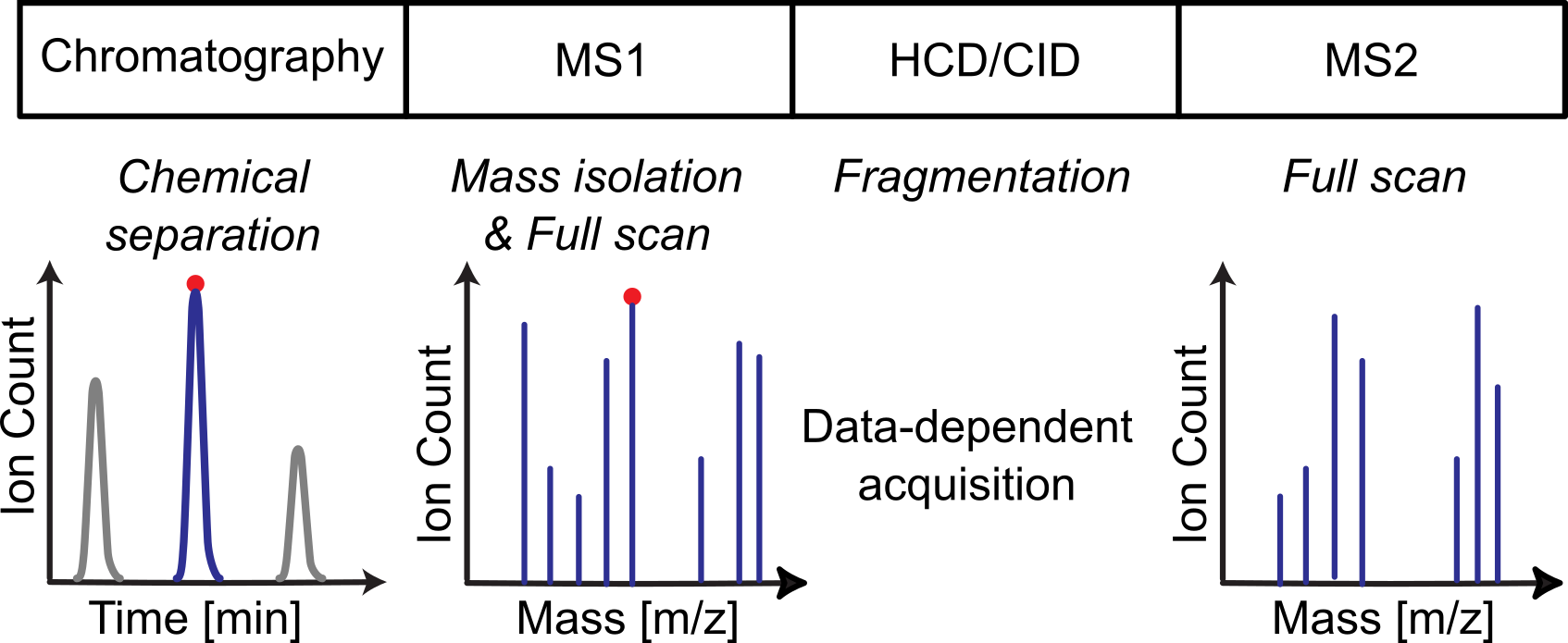
Objectives¶
Obtaining the SOP for the workflow.
Choosing a data set for demonstrating the workflow.
Creating an optimized SmartPeak input templates for running the workflow.
The Workflows include¶
Defining the accurate mass search database
Processing Unknowns based MS1 accurate mass and creating a transition library based on MS2 product ion scans
Processing Unknowns based MS1 accurate mass and creating a spectral database based on MS2 product ion scans
Processing Unknowns based on MS2 product ion scans using an existing spectral database
Reviewing the results
Notes¶
The algorithm parameters used in the following workflows have been highly tuned for feature detection using Time of Flight (Tof) technologies.
Slight modifications to the algorithm parameters in the TargetedSpectraExtractor section is needed for Orbitrap technology and other acquisition method specific parameters.
Steps¶
The tutorial includes the following steps :
Setting up the input files
The data set used can be found in LC-MS/MS-DDA for the transition library and spectral database generation, and spectral library matching workflows.
The dataset includes a
CHEMISTRYfolder which contains HMDB (Human Metabolome Database) mapping files organized as follows:HMDB2StructMapping.tsv
This tab seperated file contains mapping of HMDB IDs, IUPAC Name, Compound Summary (Synonyms), Canonical SMILES and InChl.
HMDBMappingFile.tsv
This tab seperated file contains the Monoisotopic Molecular Weight and the Chemical Formula mapped to their respective HMDB ID.
negative_adducts.tsv
This file contains negative ion modes including the charge.
positive_adducts.tsv
This file contains positive ion modes including the charge.
The above files provided in the example are appropriate for applications involving human serum and other biosamples. For applications involving other organisms such as bacteria (e.g., E. coli), the use of organism specific databases are recommended to reduce the number of false positives. See FIA_MS_database_construction.ipynb for an example notebook demonstrating how to convert the metabolites in a genome-scale reconstruction to SmartPeak accurate mass mapping files.
Furthermore, the
parameters.csvfile contains the following settings for this workflow:DDA_parameters.csv¶ function
name
value
type
FeatureFindingMetabo
report_chromatograms
TRUE
bool
FeatureFindingMetabo
report_convex_hulls
TRUE
bool
FeatureFindingMetabo
use_smoothed_intensities
FALSE
bool
Pick3DFeatures
enable_elution
FALSE
bool
Pick3DFeatures
max_traces
1000
int
Pick3DFeatures
force_processing
FALSE
bool
TargetedSpectraExtractor
AccurateMassSearchEngine:db:mapping
[‘CHEMISTRY/HMDBMappingFileGermicidinA.tsv’]
list
TargetedSpectraExtractor
AccurateMassSearchEngine:db:struct
[‘CHEMISTRY/HMDB2StructMappingGermicidinA.tsv’]
list
TargetedSpectraExtractor
AccurateMassSearchEngine:positive_adducts
CHEMISTRY/PositiveAdducts.tsv
string
TargetedSpectraExtractor
AccurateMassSearchEngine:negative_adducts
CHEMISTRY/NegativeAdducts.tsv
string
TargetedSpectraExtractor
relative_allowable_product_mass
50
float
TargetedSpectraExtractor
AccurateMassSearchEngine:ionization_mode
auto
string
TargetedSpectraExtractor
AccurateMassSearchEngine:mass_error_value
10
float
TargetedSpectraExtractor
rt_window
60
float
TargetedSpectraExtractor
mz_tolerance
0.001
float
TargetedSpectraExtractor
AccurateMassSearchEngine:keep_unidentified_masses
TRUE
bool
Defining the workflow in SmartPeak
For LC-MS/MS-DDA transition library generation analysis, the following steps are saved into the workflow.csv file.
Alternatively, steps can be replaced, added or deleted direclty from SmartPeakGUI.
A detailed explanation of each command step can be found in Workflow Commands.
workflow_LCMS_DDA_transition.csv¶ workflow_step
LOAD_RAW_DATA
PICK_3D_FEATURES
SEARCH_SPECTRUM_MS1
MERGE_FEATURES_MS1
EXTRACT_SPECTRA_NON_TARGETED
SEARCH_SPECTRUM_MS2
MERGE_FEATURES_MS2
CONSTRUCT_TRANSITIONS_LIST
STORE_FEATURES
The workflow process’s data dependent acquisition (DDA) data generated by liquid chromatography mass spectrometry instrumentation.
The workflow first picks, annotates, and merges features found in the MS1 scans for each sample, second picks, annotates, and merges features found in the MS2 scans for each sample, and third constructs a transition list for downstream quantification using e.g., data independent acquisition (DIA) or single reaction monitoring (SRM) methods.
The transition library generated for each sample can be found in the
output featuresdirectory. A snippet of the table is shown below.
DDA_transition_library.csv¶ PrecursorMz
ProductMz
PrecursorCharge
ProductCharge
LibraryIntensity
NormalizedRetentionTime
ProteinId
TransitionGroupId
TransitionId
235.0747741
134.9568506
0
NA
476
329.3195788
HMDB:HMDB0000001
HMDB:HMDB0000001
HMDB:HMDB0000001_scan=440_134.957_370.346
235.0747741
217.1050819
0
NA
464
329.3195788
HMDB:HMDB0000001
HMDB:HMDB0000001
HMDB:HMDB0000001_scan=440_217.105_370.346
Statistics on each of the individual scans analyzed can be viewed at
view | scans.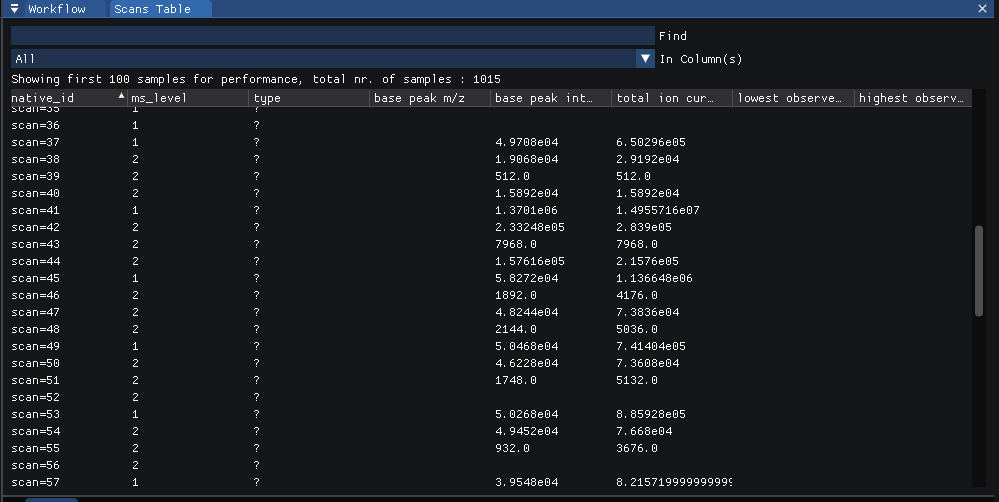
The features found can be viewed at
view | features (table)in long form.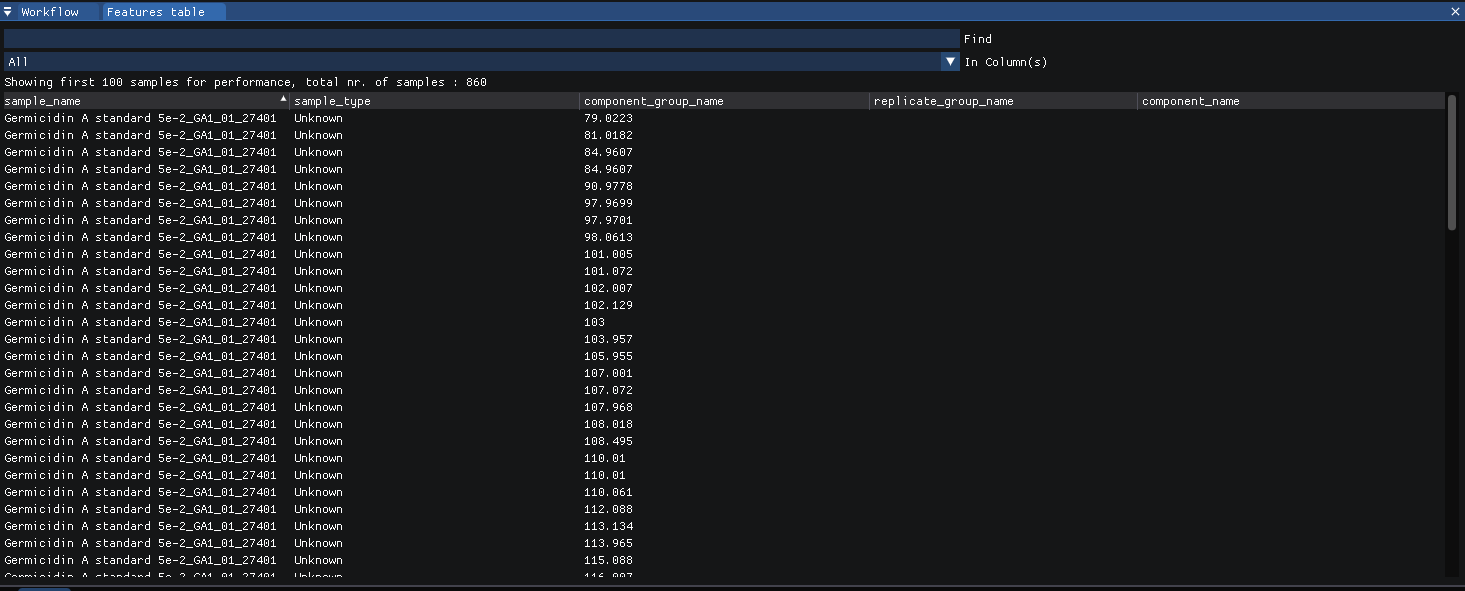
Or at
view | features (matrix)in compact form.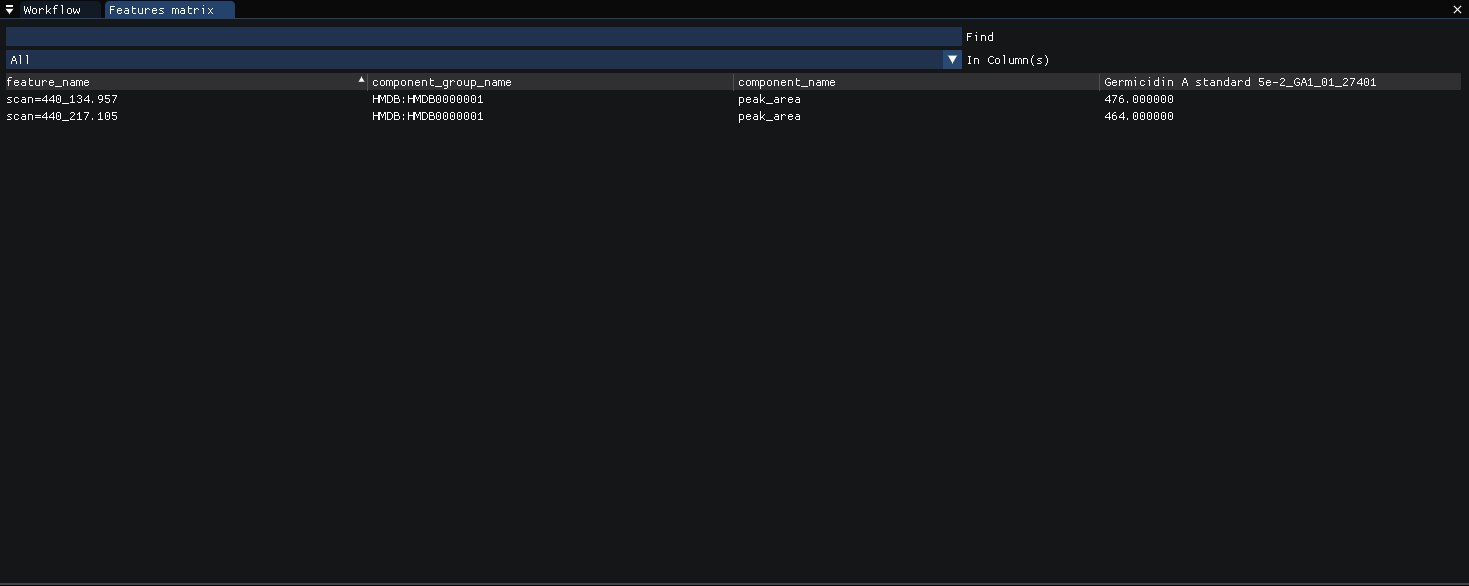
The TIC for each injection can be viewed at
view | chromatograms.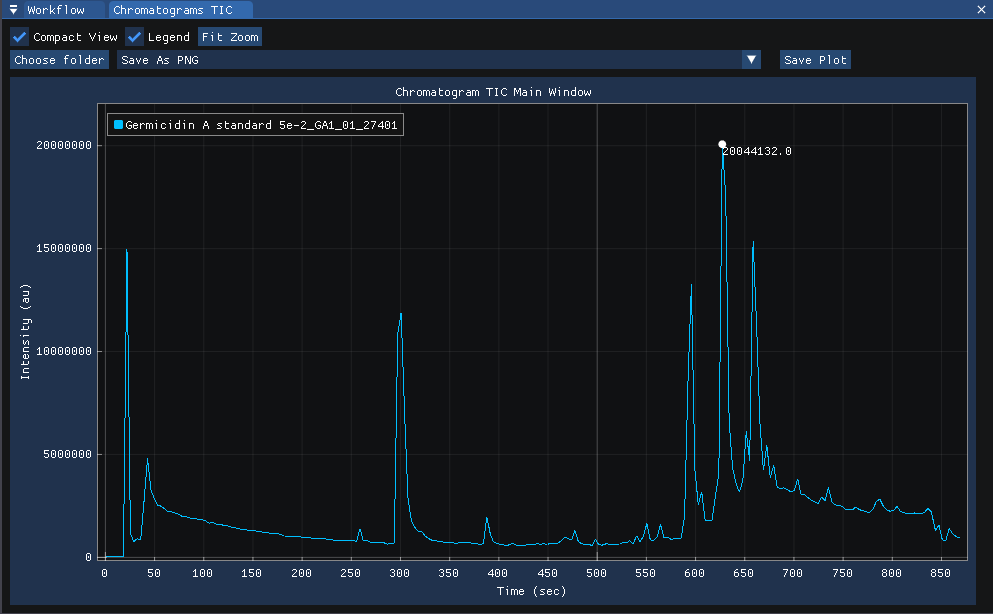
Clicking on the chromatogram reveals the MS1 spectra acquired at the selected time-point.
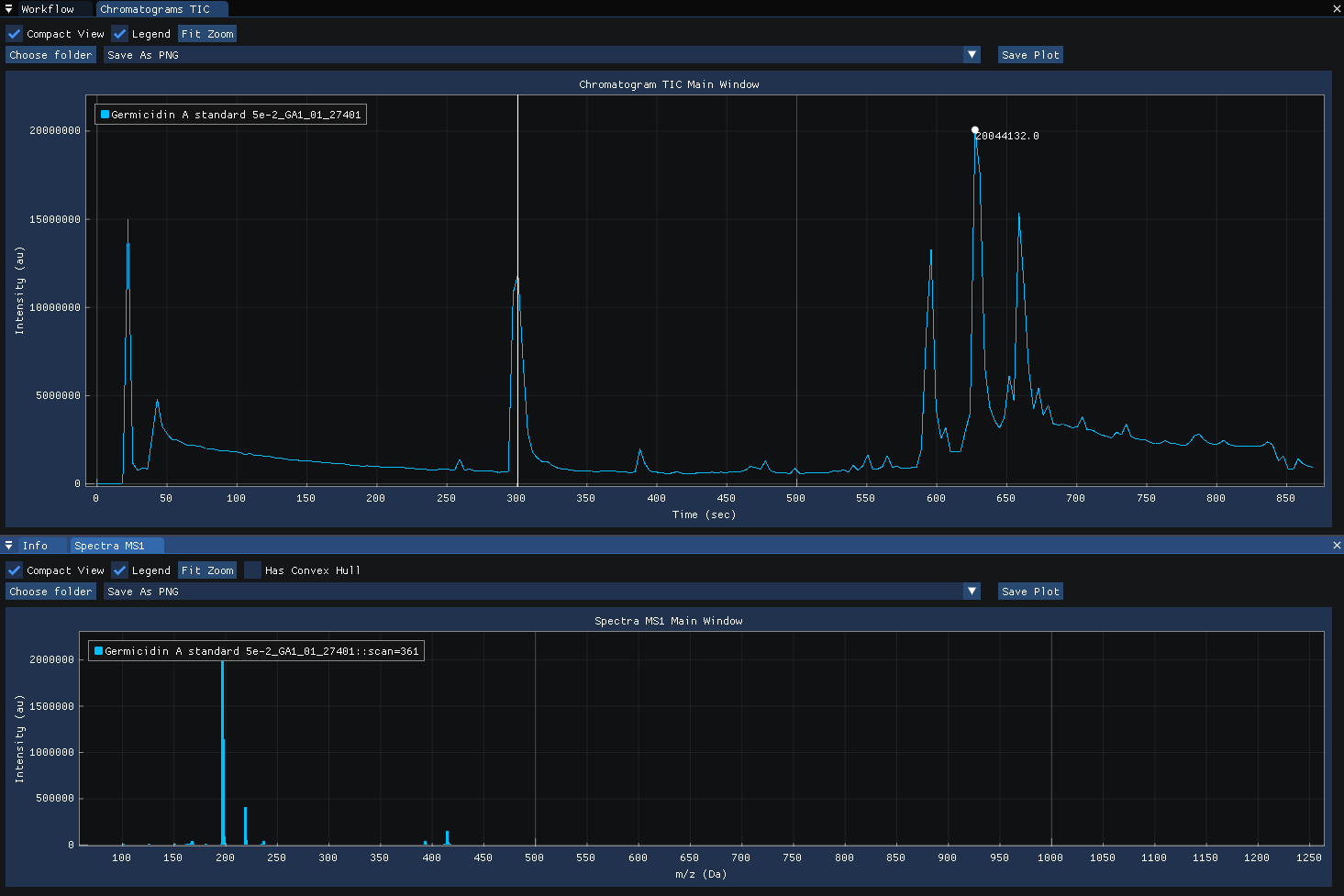
Clicking on the spectra computes and displays the MS1 XIC for the selected m/z.
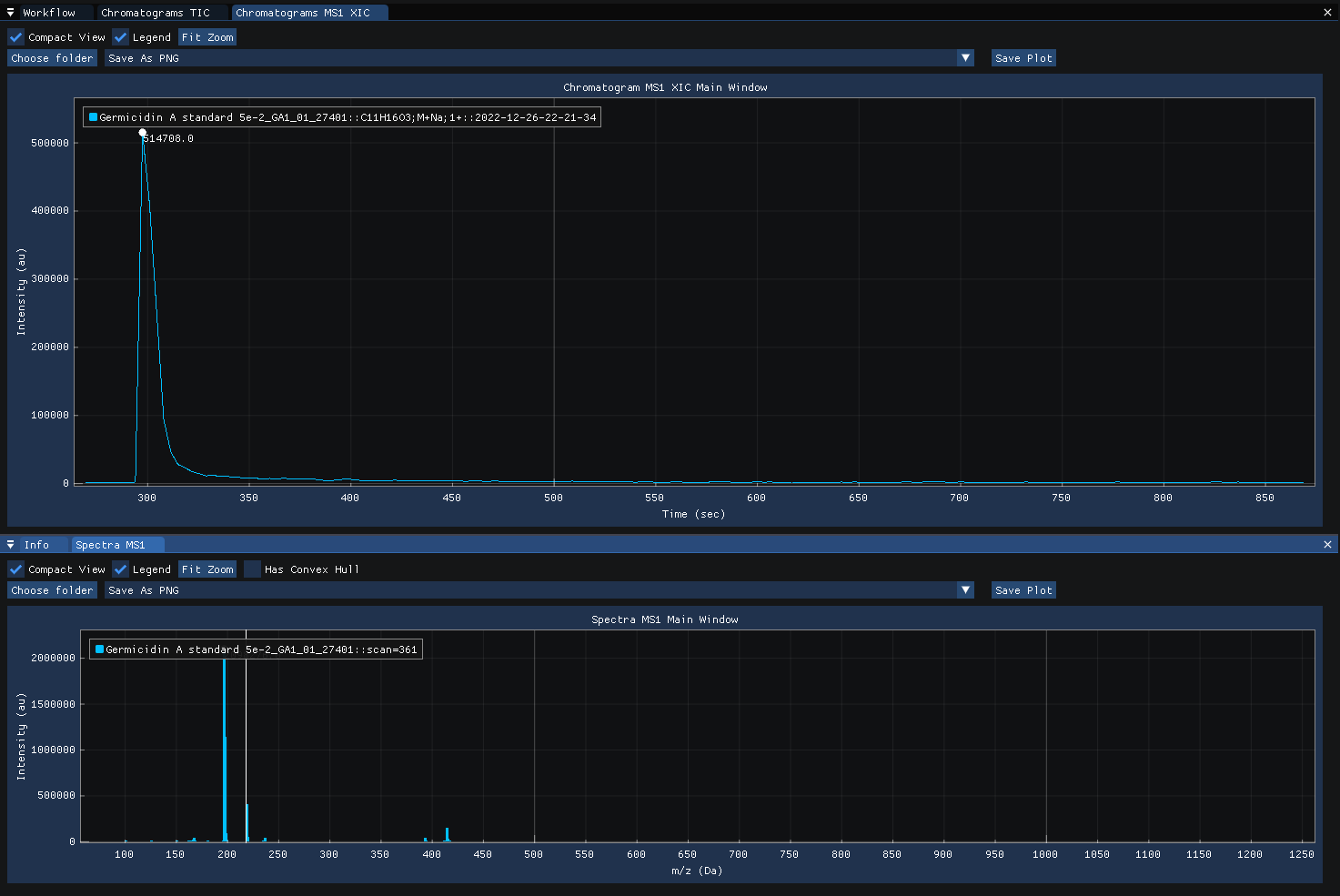
Clicking on the MS1 XIC chromatogram revealse the MS2 spectra acquired at the selected time-point.
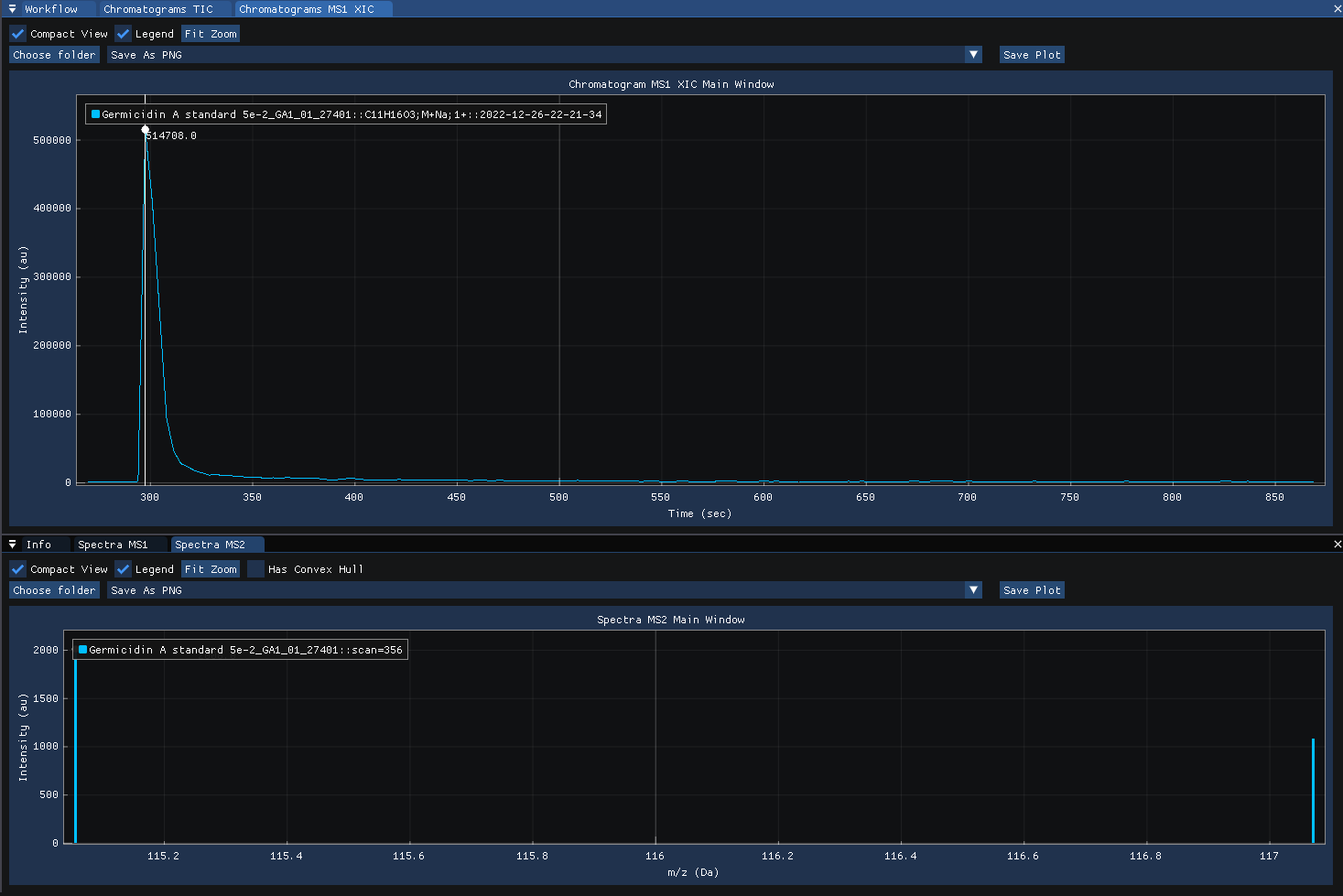
Defining the workflow in SmartPeak
For LC-MS/MS-DDA spectral database generation analysis, the following steps are saved into the workflow.csv file.
Alternatively, steps can be replaced, added or deleted direclty from SmartPeakGUI.
A detailed explanation of each command step can be found in Workflow Commands.
workflow_LCMS_DDA_spectral.csv¶ workflow_step
LOAD_RAW_DATA
PICK_3D_FEATURES
SEARCH_SPECTRUM_MS1
MERGE_FEATURES_MS1
EXTRACT_SPECTRA_NON_TARGETED
STORE_MSP
STORE_FEATURES
The workflow process’s data dependent acquisition (DDA) data generated by liquid chromatography mass spectrometry instrumentation.
The workflow first picks, annotates, and merges features found in the MS1 scans for each sample, second picks features found in the MS2 scans for each sample, and third constructs a spectral library for downstream spectral annotation.
The spectral database in MSP format generated for each sample can be found in the
output featuresdirectory. A snippet of the table is shown below.
DDA_spectral_library.csv¶ Name: HMDB:HMDB0000001
Retention Time: 370.346
base peak intensity: 476.0
total ion current: 940.0
Num Peaks: 2
134.957:476 217.105:464
Defining the workflow in SmartPeak
For LC-MS/MS-DDA spectral database matching analysis, the following steps are saved into the workflow.csv file.
Alternatively, steps can be replaced, added or deleted direclty from SmartPeakGUI.
A detailed explanation of each command step can be found in Workflow Commands.
workflow_LCMS_DDA_spectra.csv¶ workflow_step
LOAD_RAW_DATA
PICK_3D_FEATURES
EXTRACT_SPECTRA_NON_TARGETED
MATCH_SPECTRA
STORE_FEATURES
The workflow process’s data dependent acquisition (DDA) data generated by liquid chromatography mass spectrometry instrumentation.
The workflow first picks features found in the MS1 scans for each sample, second picks features found in the MS2 scans for each sample, and third annotates the MS1 features by matching the spectra of the MS2 scans with a spectral library.