Targeted flux analysis using GC-MS full-scan acquisition¶
This tutorial walks you through the workflow for analyzing targeted full-scan flux analysis using SIM GC-MS data starting from input file generation, to processing the data in SmartPeak, to reviewing the data in SmartPeak, to reporting the results.
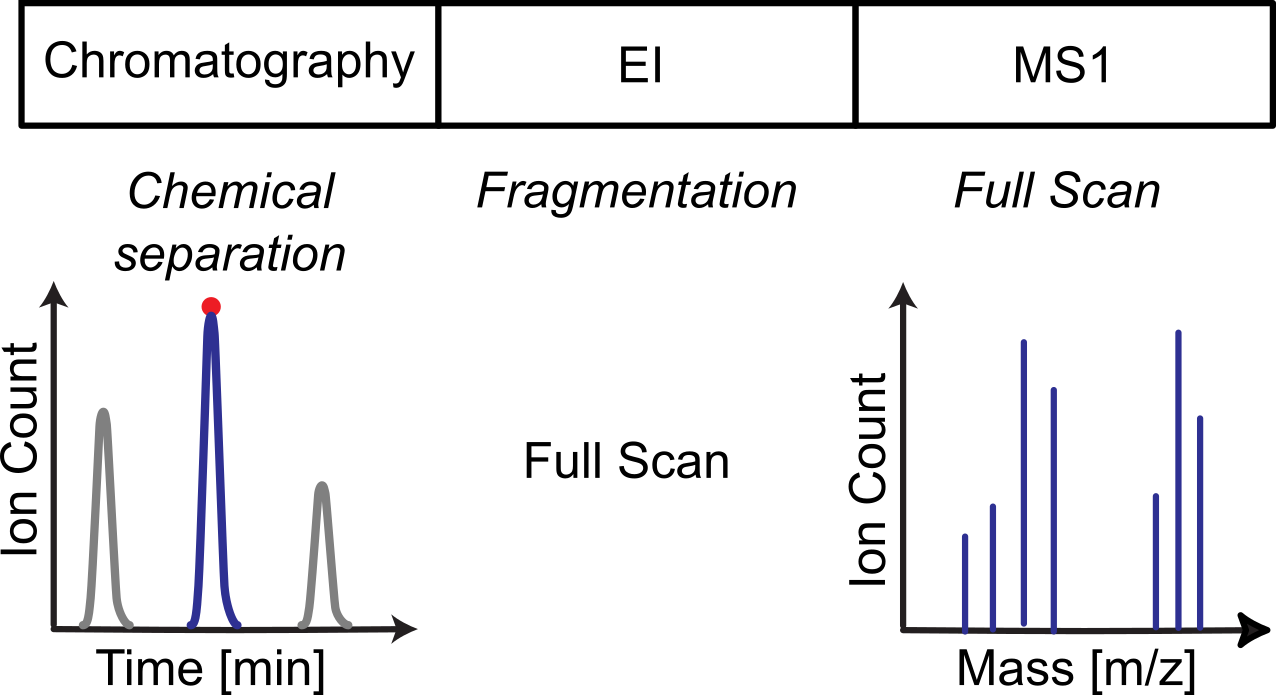
Objectives¶
Obtaining the SOP for the workflow.
Choosing a data set for demonstrating the workflow.
Creating an optimized SmartPeak input templates for running the workflow.
The Workflows include¶
Processing Unknowns
Reviewing the results
Notes¶
The algorithm parameters used in the following workflows have been highly tuned for feature detection using an Agilent GC and single quad system. With that said, we have found the algorithm parameters to generalize well to most gas chromatography coupled to mass spectrometry systems.
Steps¶
The tutorial includes the following steps :
Setting up the input files
The data set used can be found here ProAAs GCMS Fullscan.
The dataset includes a
parameters.csvfile containing parameter settings forMRMFeatureFinderScoring,MRMFeatureSelector,MRMFeatureValidator,ReferenceDataMethods,MRMFeatureSelector,MRMMapping,CalulateMDVs,IsotopicCorrectionsandCalculateMDVAccuracies.Defining the workflow in SmartPeak
For the targeted flux analysis via SIM GCMS, the following steps are saved
into the workflow.csv file. Alternatively, steps can be replaced,
added or deleted direclty from SmartPeakGUI within the “workflow” tap in the right pane.
A detailed explanation of each command step
can be found in Workflow Commands.
workflow_GCMSFullScan_Flux_Unknowns.csv¶ workflow_step
LOAD_RAW_DATA
MAP_CHROMATOGRAMS
EXTRACT_CHROMATOGRAM_WINDOWS
ZERO_CHROMATOGRAM_BASELINE
PICK_MRM_FEATURES
QUANTIFY_FEATURES
CHECK_FEATURES
SELECT_FEATURES
CALCULATE_MDVS
ISOTOPIC_CORRECTIONS
CALCULATE_MDV_ACCURACIES
STORE_FEATURES
The workflow pipeline is initialized by loading the raw data followed by mapping the chromatograms to the loaded set of transitions. Once done, chromatogram windows will be extracted based on the componentFeatureFilter parameters, then for normalizing the lowest chromatogram intensity to zero. The next step in the pipeline is to execute the peak picking routine on the SRM/MRM features followed by applying a calibration model to each transition as defined in the quantitationMethods parameters. Transitions and transition groups are flagged and scored per user pre-defined criteria, then the peak selection/ alignment algorithms is executed. Mass distribution vectors can then be calculated followed by isotopic corrections and comparing the MDVs to the theoretical values. As the final step in the workflow, the features for the sample group is stored to disk as a
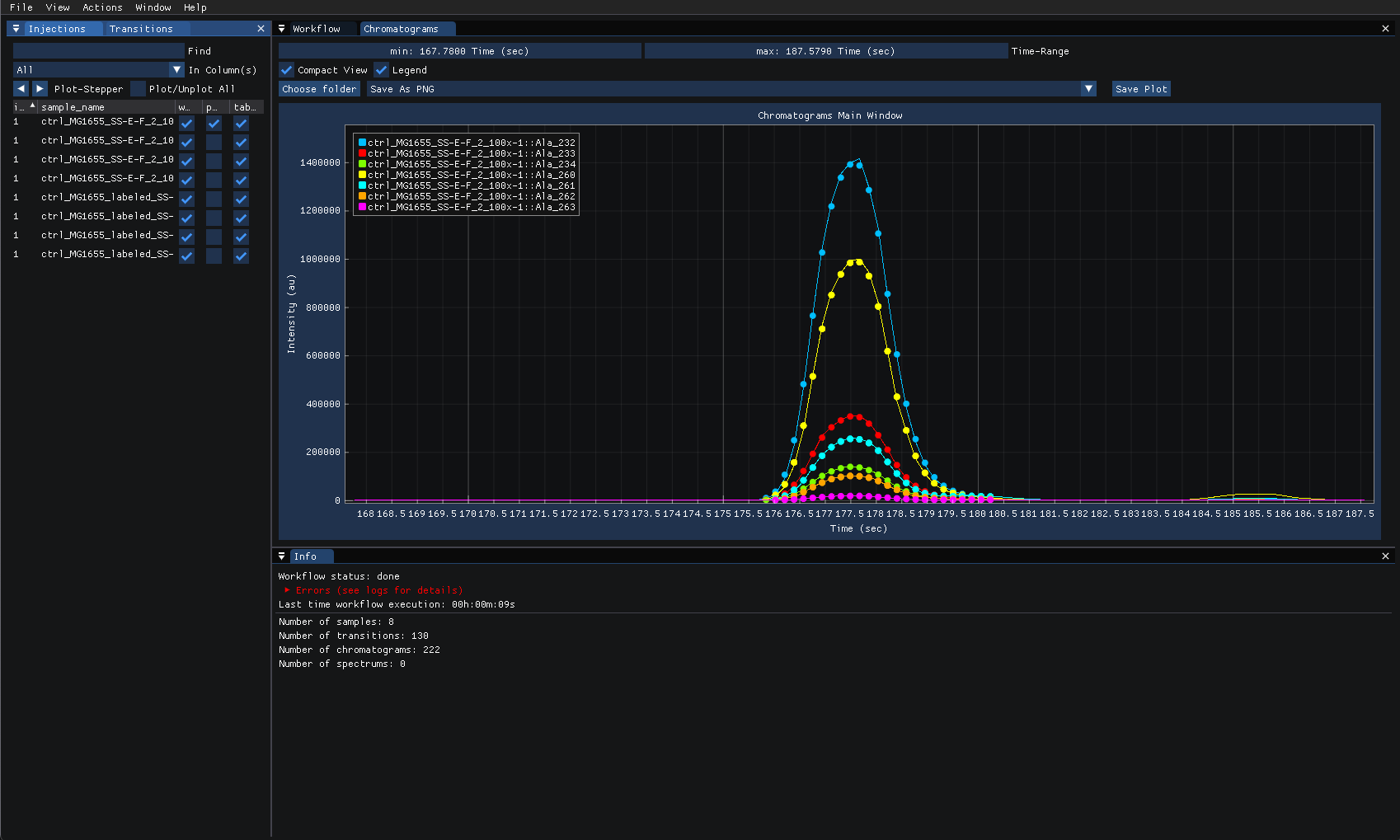
featureXMLfiles.To plot the intensities over time for given injections and transitions, view the “chromatogram” from the “view” menu then select the injections and transitions to plot from their respective tabs on the left. The following shows the chromatogram for one injection using Ala_232 transition and their intensity differences over time.
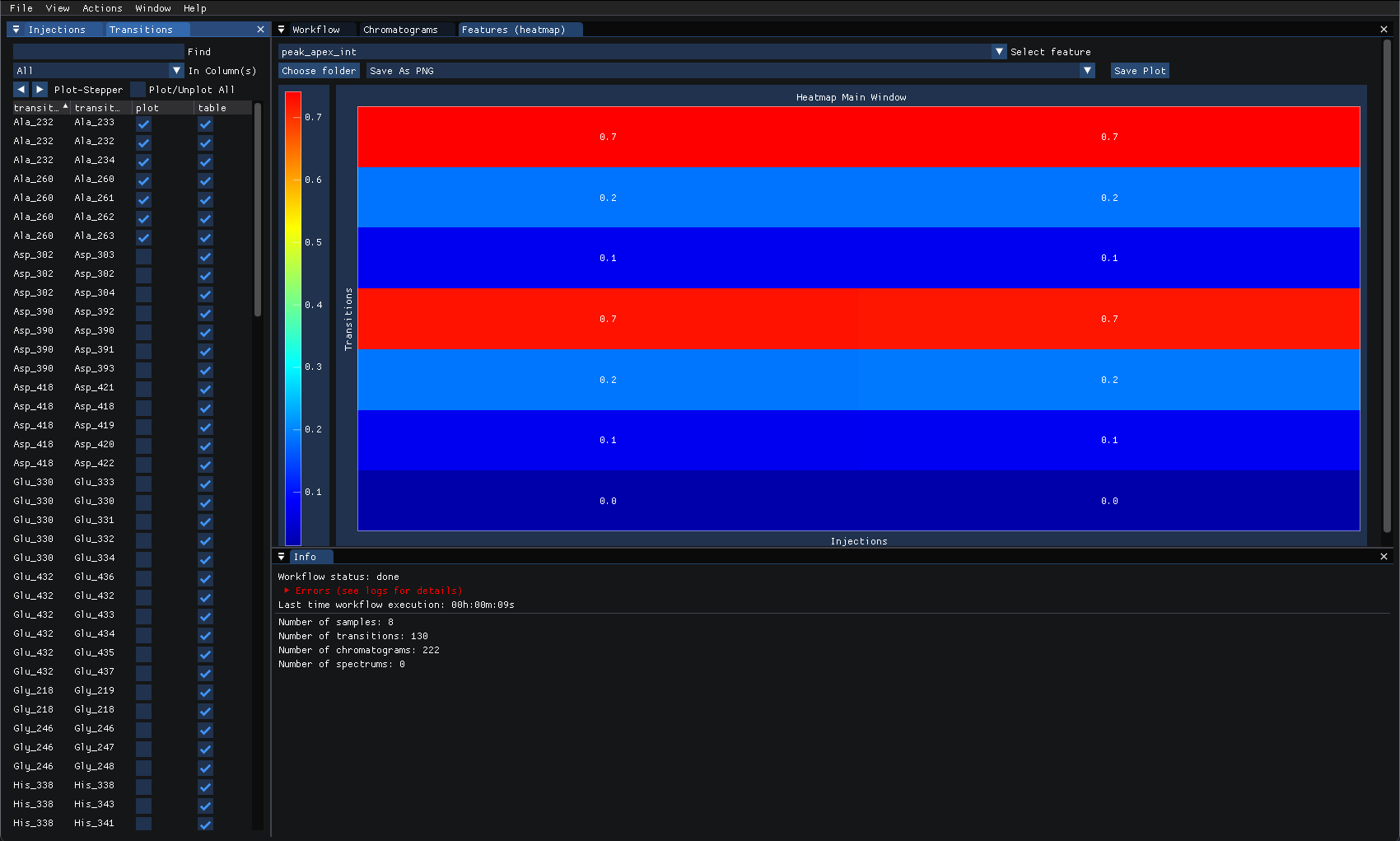
The features can also be plotted as a heatmap, under “view” select “Features (heatmap)” then select the “peak_apex_int” feature to display transition groups as a heatmap and compare the values from the same injection as shown below:
Reporting the results
To export the results, select “Report” from the “Actions” which will show the “Create Report” window:
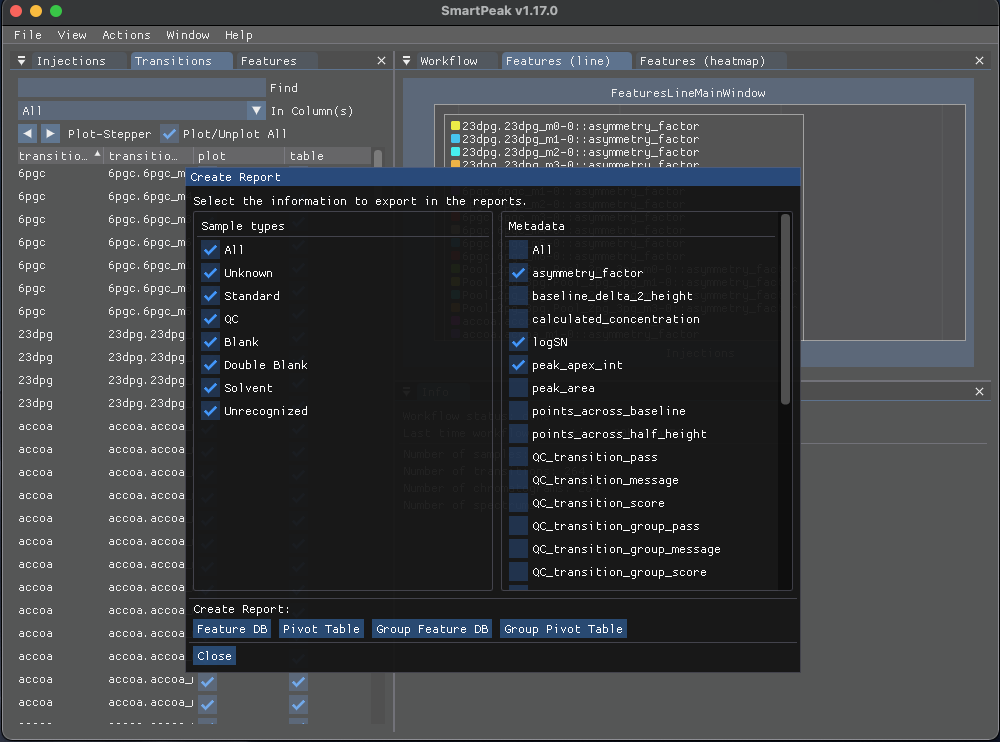
Based in the data you wish to export, select the desired “Sample types” from the left pane and select the “Metadata” from the right pane then click on one of the buttons below to create the report with the selected items in the csv format.
More details on exporting the results can be found in Export report.
The resulting
featureXMLandmzTabare saved in the features folder.featureXMLfiles describe the spectra data for a given injection sample which includes a list of features with a set ofUserParamfor each feature such asPeptideRef,native_idandscan_polarity.